Converting GFF3 to refFlat
- By: Faith Okamoto
- Original development: April 2023
- Writeup: July 2024
Full copy-paste-able code is right before References.
Introduction
Palmer Lab performs genome-wide association studies (GWAS) to determine genetic drivers of complex traits such as addiction. GWAS identify regions of the genome associated with the given phenotype. Association significance is assessed via p-values. However, due to local linkage disequilibrium (LD), i.e. the tendency for physically close variants to be inherited as a unit without recombination, many variants have similar p-values.
To identify the causal signal in a quantitative trait locus (QTL), we visualize GWAS results in a regional association plot (e.g. Gunturkun et al. 2022). These display the association signal in the context of local genes. Genes within or nearby the block of associated variants are more likely to be driving the association signal. These genes can then be prioritized for follow-up analyses.
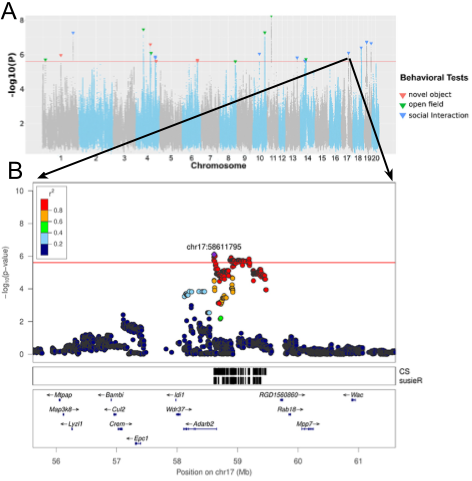
Figure 1. Example plots from Gunturkun et al. 2022. A. Porcupine plot with genotype-phenotype associations for all phenotypes in the study. Variant position (separated by chromosome) on X-axis, significance level on Y-axis. Taken from Figure 3. B. Regional association plot for "latency of entering social zone in SIT" QTL on Chromosome 17. Note the included gene annotations. Taken from Figure 6.
Regional association plots require the positions of genes to plot. We use the refFlat format, as required by LocusZoom. However, the Rat Genome Database (RGD) releases genome annotations in GFF3 format. Thus, we need to convert from one format to the other.
File formats
Note that for annotations on the - strand, start and end are flipped. For example, txStart is the end of transcription. This ensures that end > start.
GFF3
GFF3 is a tab-separated nine-column format. It starts with #-marked comments, which provide metadata such as format version. Column headers are not provided.
| Col # | Used by script | Column name | Example | Description |
|---|---|---|---|---|
| 1 | Yes | seqid | Chr1 | chromosome/contig name |
| 2 | No | source | RGD | annotation source |
| 3 | Yes | type | exon | type of annotation |
| 4 | Yes | start | 53396 | 1-indexed start position |
| 5 | Yes | end | 53701 | 1-indexed end position |
| 6 | No | score | . | floating point value |
| 7 | Yes | strand | - | annotation strand |
| 8 | No | phase | 0 | 0-indexed codon start position |
| 9 | Yes | attributes | Parent=mRNARGD11369141;ID=trFeature11369144 | tag-value pairs |
Only the Name (genes only), Parent (non-genes only), and ID attributes are used. Each gene has its own line, and then each of its mRNAs, exons and coding sequences (CDS) has its own line as well.
These annotations are taken from RGD
RefFlat
RefFlat is a tab-separated 11-column format. It starts with a header row. The following table is modified from the UCSC Genome Browser.
| Col # | Field | Example | Description |
|---|---|---|---|
| 1 | geneName | PSRC1 | Common name of gene |
| 2 | name | NM_001283145 | Gene identifier |
| 3 | chrom | chr1 | chromosome/contig name |
| 4 | strand | + | annotation strand |
| 5 | txStart | 117473117 | 0-indexed transcription start position |
| 6 | txEnd | 117476379 | 0-indexed transcription end position |
| 7 | cdsStart | 117473521 | 0-indexed coding region start position |
| 8 | cdsEnd | 117476075 | 0-indexed coding region end position |
| 9 | exonCount | 8 | Number of exons |
| 10 | exonStarts | 117473117,117473483,1174736... | 0-indexed exon start positions |
| 11 | exonEnds | 117473147,117473540,1174737... | 0-indexed exon end positions |
Each gene and all of its exons are on the same line.
Conversion method
- Read raw file into
gff3: the GFF3 is read (ignoring header comments). The two core attributes (IDandParent) are extracted from theATTRcolumn and put into their own columns. - Clean up
gff3: minor tweaks to make the GFF3 closer to refFlat data.- In a GFF3, exons/CDS which are part of an mRNA have the mRNA as their
Parent. In a refFlat, exons are directly connected to their parent gene. Thus, theParentof each mRNA is used to overwrite its children's. - Many of the lines in a GFF3 are not needed in a refFlat. These include
three_prime_UTR,five_prime_UTR,mRNA, and any sort of gene with aParent(since refFlat needs genes to be the top-level unit). - Some genes aren't given a separate exon/CDS row in the GFF3. Since refFlat needs exon annotations, these are given a dummy whole-gene exon.
- In a GFF3, exons/CDS which are part of an mRNA have the mRNA as their
- Reformat into refFlat: for each gene in
gff3:- Copy data over to form the first six
refFlatcolumns. - Take all exons, sort by start coordinate, merge overlapping exons (i.e. use maximum edges for alternative splicing), and distill into
exonCount,exonStarts, andexonEndscolumns. - Take most extreme CDS positions for the
cdsStartandcdsEndcolumns.
- Copy data over to form the first six
- Save
refFlat: combine Step 3 by-gene information and save to file.
Code
This code requires the package data.table. It was tested for data.table version 1.14.8 and R version 4.2.3.
library(data.table)
# ---- read gff3 basics ----
# https://useast.ensembl.org/info/website/upload/gff3.html
gff3 <- fread(<infile>, header = FALSE,
col.names = c("SEQID", "SOURCE", "TYPE", "START", "END",
"SCORE", "STRAND", "PHASE", "ATTR"))
# extract <val> from [anything];<name>=<val>;[anything]
extract_attr <- function(name, x) {
ifelse(grepl(name, x), gsub(paste0(".*;", name, "=*(.*?);.*"), "\\1", x), NA)
}
# the extract_attr() function expects everything to be bordered by semicolons
gff3[, ATTR := paste0(";", ATTR, ";")]
# basic attributes which are needed for multiple purposes; extracting them once
gff3[, ID := extract_attr("ID", ATTR)][, PARENT := extract_attr("Parent", ATTR)]
# ---- clean up gff3 ----
# for chains of feature -> mRNA -> gene, create a pointer from feature -> gene
mRNAs <- gff3[TYPE == "mRNA"]
mRNAs[, PARENT_PARENT := PARENT][, PARENT := NULL]
gff3 <- merge.data.table(gff3, mRNAs[, c("ID", "PARENT_PARENT")],
by.x = "PARENT", by.y = "ID", all.x = TRUE)
gff3[!is.na(PARENT_PARENT), PARENT := PARENT_PARENT][, PARENT_PARENT := NULL]
# we don't care about these separately
gff3 <- gff3[!(TYPE %in% c("three_prime_UTR", "five_prime_UTR", "mRNA"))]
# these are used as filler single-exon things when no exon is known
types_of_genes <- c("pseudogene", "protein_coding_gene",
"ncRNA_gene", "tRNA_gene", "gene")
gff3 <- gff3[!(TYPE %in% types_of_genes & !is.na(PARENT))]
# some genes lack exons or CDS; pretend these are exons and CDS unto themselves
childless_genes <- gff3[TYPE == "gene" & !(ID %in% PARENT)]
gff3 <- rbindlist(list(gff3,
copy(childless_genes)[, TYPE := "exon"][, PARENT := ID],
copy(childless_genes)[, TYPE := "CDS"][, PARENT := ID]))
# ---- extract information for refFlat file ----
# get basic information about each gene
refFlat <- gff3[TYPE == "gene",
.(geneName = extract_attr("Name", ATTR), name = ID,
chrom = paste0("c", substring(SEQID, 2)), strand = STRAND,
txStart = START, txEnd = END)]
# helper function to determine unique exons given starts and ends
get_exons <- function(starts, ends) {
exons <- data.table(START = starts, END = ends)
exons <- exons[!duplicated(exons)][order(START)]
# merge overlapping exons
exons <- exons[, .(START = min(START), END = max(END)),
by = .(group = cumsum(c(1, tail(START, -1) > head(END, -1))))]
# extract number and ends of exons
return(list(exonCount = nrow(exons),
exonStarts = paste(exons$START, collapse = ","),
exonEnds = paste(exons$END, collapse = ",")))
}
# find exons & coding-sequence boundaries by gene
exons <- gff3[TYPE == "exon", get_exons(START, END), by = PARENT]
cds <- gff3[TYPE == "CDS", .(cdsStart = min(START), cdsEnd = max(END)),
by = PARENT]
# --- combine into final refFlat ----
# add these columns to the refFlat
refFlat <- merge.data.table(refFlat, merge.data.table(exons, cds),
by.x = "name", by.y = "PARENT")
# https://genome.sph.umich.edu/wiki/LocusZoom_Standalone#Inserting_refFlat
setcolorder(refFlat, c("geneName", "name", "chrom", "strand", "txStart",
"txEnd", "cdsStart", "cdsEnd", "exonCount",
"exonStarts", "exonEnds"))
write.table(refFlat, <outfile>,
quote = FALSE, sep = "\t", row.names = FALSE)
References
Gunturkun MH, Wang T, Chitre AS, Garcia Martinez A, Holl K, St. Pierre C, Bimschleger H, Gao J, Cheng R, Polesskaya O, et al. 2022. Genome-Wide Association Study on Three Behaviors Tested in an Open Field in Heterogeneous Stock Rats Identifies Multiple Loci Implicated in Psychiatric Disorders. Front Psychiatry. 13. doi:10.3389/fpsyt.2022.790566.

Comments are closed.